近日,我校催化反应工程团队段学志教授、曹约强特聘研究员等在数据驱动的高性能低碳炔烃选择性加氢催化剂设计与创制方面取得新进展,基于机器学习构建的催化剂高通量筛选模型,快速筛选能够在原子尺度精准调控Ni活性位点结构的客体金属,实现了关键物种构型匹配以及目标反应路径的定向调控。相关成果以“Atomic Design of Alkyne Semihydrogenation Catalysts via Active Learning”为题在线发表在《美国化学会志》(Journal of the American Chemical Society)上。
石脑油裂解碳二和碳三馏分中少量的炔烃选择性加氢是制备聚合级乙烯和丙烯的关键。非贵金属Ni因其具有未填充的d轨道电子而表现出优异的氢活化能力,然而该类催化剂选择性往往较低。研究团队先前的研究结果表明,设计和制备原子级分布的金属活性位点、通过引入第二金属精确调控其上关键物种的吸附构型能实现对目标产物选择性的调控(Angew. Chem. Int. Ed. 2020, 59, 11647; Angew. Chem. Int. Ed. 2022, 61, e202215225; Nat. Commun. 2022, 13, 5534)。为寻找最优的第二金属,传统研究模式主要依赖实验试错方法,效率较低。快速发展的机器学习等数据科学技术为催化剂设计带来新的契机。基于机器学习、理论计算和实验研究的有机结合,有望实现催化剂的高效筛选,突破传统试错法的弊端。
为此,研究团队提出了基于贝叶斯优化的主动学习框架与DFT计算相结合的研究方法,以乙烯脱附和其进一步加氢的能垒差值作为选择性描述符,构建了自动化催化剂高通量筛选的工作流程,用于预测乙炔选择性加氢的高性能Ni基金属间化合物。随后,从3000多个候选Ni基金属间化合物中快速筛选出15个高性能Ni基金属间化合物作为潜在的炔烃加氢催化剂,利用DFT计算进一步验证了ML模型的预测精度,最终确定了所推荐的NiIn催化剂为最优候选催化剂供实验进一步验证。
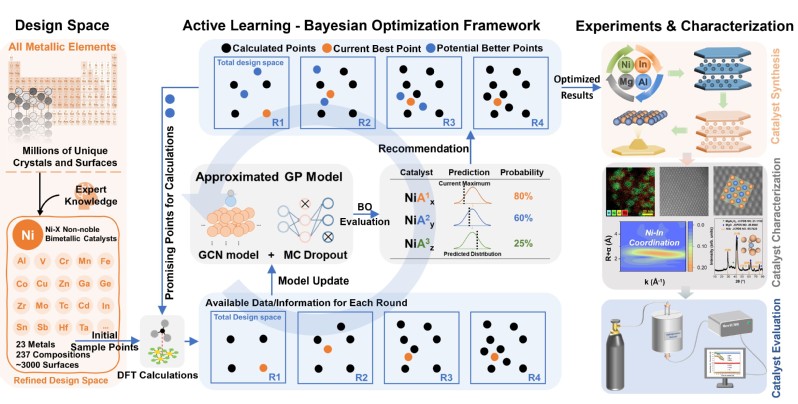
研究团队进一步通过实验合成NiIn金属间化合物催化剂及参比Ni基催化剂,并借助球差电镜和X射线吸收光谱等表征技术解析了催化剂结构,证明了长程有序、结构稳定的NiIn金属间化合物催化剂的成功制备。催化反应性能评价表明:NiIn金属间化合物催化剂在乙炔和丙炔转化率为100%时,乙烯和丙烯选择性高达97.0%,明显高于参比催化剂,证实了理论计算的预测结果。结合吸附构型分析、电子结构分析、原位红外光谱表征等机理研究手段,发现NiIn催化剂优异性能主要归因于Ni 3d与In 5s-p轨道的能量匹配和适宜的杂化,尤其是Ni 3d与In 5p轨道的杂化。

该论文第一作者为葛小虎博士、新加坡国立大学博士研究生殷骏和上海交通大学博士研究生任洲宏,通讯作者为威尼斯886699催化反应工程团队段学志教授、曹约强特聘研究员、清华大学王笑楠教授和上海交通大学刘晰教授。论文研究工作得到了袁渭康院士和周兴贵教授的悉心指导。此外,该研究工作得到了国家重点研发计划等项目的支持。
原文链接:https://pubs.acs.org/doi/full/10.1021/jacs.3c14495